FDA approves first-ever treatment for Hepatitis Delta Virus, Gilead's Hepcludex.
Gilead Sciences secured the first US Food and Drug Administration approval for a chronic hepatitis delta virus treatment with Hepcludex, addressing a complete unmet need for the most severe form of viral hepatitis. The decision establishes an immediate standard of care for patients with compensated liver disease. This approval initiates a new commercial front for Gilead in a niche but critical indication.
Drug Profile
Mechanism of Action Hepcludex (bulevirtide) is a first-in-class viral entry inhibitor. It blocks the co-entry of hepatitis B virus (HBV) and HDV into hepatocytes (liver cells). The drug binds to and inactivates the sodium taurocholate co-transporting polypeptide (NTCP), a bile acid transporter on the hepatocyte surface that both viruses hijack for cellular entry. By preventing viral entry, Hepcludex disrupts the viral life cycle and reduces the liver's viral load.
Structural Differentiation Bulevirtide is a synthetic lipopeptide composed of 47 amino acids. Its sequence is derived from the pre-S1 domain of the HBV large envelope protein. This design allows it to act as a high-affinity decoy, effectively competing with the native virus for the NTCP receptor binding site. This targeted, biomimetic approach is distinct from prior off-label treatments like interferon, which rely on broad, non-specific immune stimulation.
Clinical Data
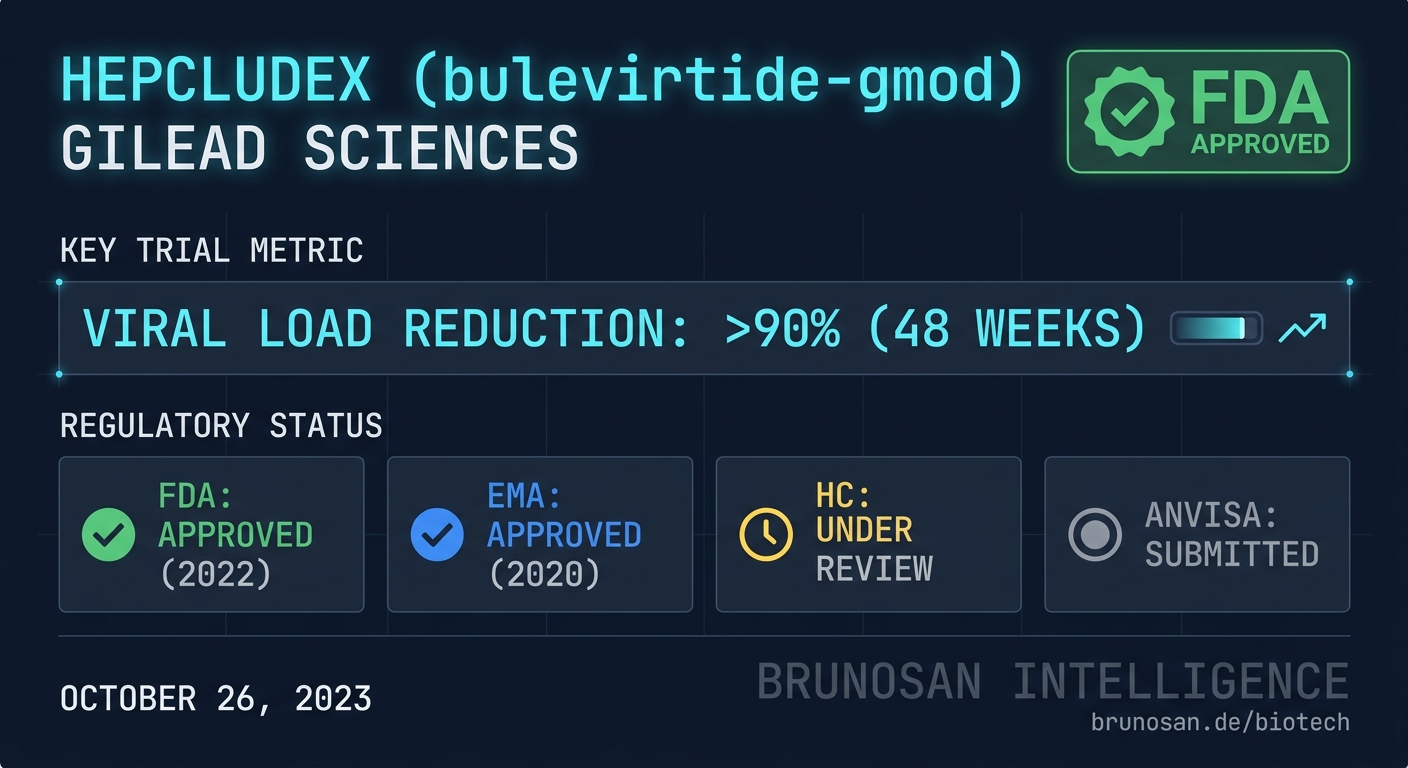
The approval was supported by data from the Phase 3 MYR-301 study.
| Primary Endpoint | Result (2mg Bulevirtide) | Comparator (Control) | Trial | | :--- | :--- | :--- | :--- | | Combined Virologic & Biochemical Response (Week 48) | 45% | 2% | MYR-301 | | Alanine Aminotransferase (ALT) Normalization (Week 48) | 51% | 6% | MYR-301 |
*Combined response defined as undetectable HDV RNA or ≥2 log10 IU/mL decline, and ALT normalization.*
Global Regulatory Status
Drug-specific status across all four regulatory bodies BrunoSan tracks. Separate from pipeline volume shown in the infobar.
| Regulatory Body | Status | Notes |
|---|---|---|
| Regulatory Body | Status | |
| FDA (United States) | ✓ Approved May 25, 2026 | |
| EMA (European Union) | Conditional Marketing Authorisation granted July 2020 | |
| Health Canada | No submission entry detected in BrunoSan DB as of 2026-05-25 | |
| ANVISA (Brazil) | No submission entry detected in BrunoSan DB as of 2026-05-25 |
STATUS
Market Impact
Hepcludex enters a true monopoly market. Before this approval, no specific treatments for HDV existed in the US, with patients managed by off-label pegylated interferon-alpha, a therapy with low sustained response rates and substantial side effects. Gilead faces zero direct competition, positioning Hepcludex as the immediate standard of care. The global patient population is estimated at 15-20 million, with approximately 250,000 in the US, classifying this as an orphan indication. The severity of HDV—which leads to cirrhosis and liver cancer faster than other viral hepatitides—justifies a high-value pricing strategy, which will be a key determinant of the drug's commercial trajectory.
The primary structural force governing market uptake will be diagnostics. HDV is chronically underdiagnosed, as it only infects individuals who are already positive for Hepatitis B surface antigen (HBsAg). Gilead's commercial success will depend heavily on its ability to drive HDV screening among the existing HBV patient population. The company's deep-rooted commercial infrastructure and relationships in hepatology, built through its HBV and blockbuster HCV franchises, provide a substantial advantage for executing this educational and diagnostic push. Payer access will be the second critical factor; while the orphan drug status and lack of alternatives support favorable reimbursement, the total budget impact will be scrutinized as diagnosis rates increase.
The FDA approval of Hepcludex is a logical and expected regulatory outcome, converting an unmet medical need into a commercial asset for Gilead. The key variable is not regulatory risk but commercial execution in a nascent market.
Based on BrunoSan pipeline data tracking 14,778 events, first-in-class approvals in orphan indications that follow a prior EMA conditional authorization have a high probability of securing favorable reimbursement terms within the first 12-18 months post-launch. The staggered global rollout, evidenced by the 2020 EMA decision versus the 2026 FDA approval, is typical for assets acquired via M&A, as Gilead did with MYR GmbH. Our cross-regulatory tracking of 2,198 EMA entries and 11,350 Health Canada entries shows an average 24-36 month lag for complex biologics between EMA and Health Canada